Understanding the role of potential biomarkers in attenuating multiple sclerosis progression via multiomics and network-based approach
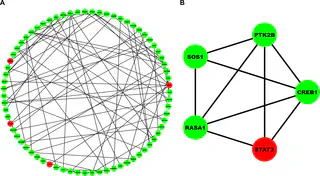 Image credit: journals
Image credit: journalsAbstract
We procured differentially expressed genes in MS patients and healthy controls by accessing mRNA dataset from a publicly accessible database. The DEGs were subjected to a non-trait weighted gene co-expression network (WGCN) for hub DEGs identification. These hub DEGs were utilized for enrichment, protein-protein interaction network (PPIN), and feed-forward loop (FFL) analyses. We identified 880 MS-associated DEGs. WGCN revealed a total of 122 hub DEGs of which most significant pathway, gene ontology (GO)-biological process (BP), GO-molecular function (MF) and GO-cellular compartment (CC) terms were assembly and cell surface presentation of N-methyl-D-aspartate (NMDA) receptors, regulation of catabolic process, NAD(P)H oxidase H2O2 forming activity, postsynaptic recycling endosome. The intersection of top 10 significant pathways, GO-BP, GO-MF, GO-CC terms, and PPIN top cluster genests identified STAT3 and CREB1 as key biomarkers. Based on essential centrality measures, CREB1 was retained as the final biomarker. Highest-order subnetwork FFL motif comprised one TF (KLF7), one miRNA (miR-328-3p), and one mRNA (CREB1) based on essential centrality measures.